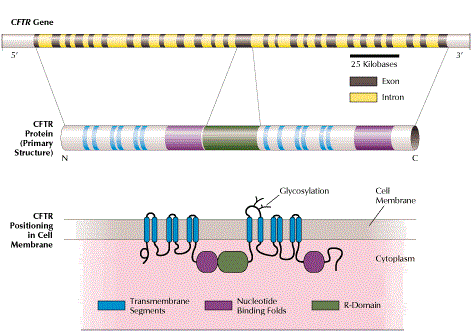
Cystic fibrosis (CF) is caused by over 1800 different mutations of the CFTR gene and is the most common genetic disease in the Caucasian population, with a carrier frequency of 1/25 – 1/40.
The main characteristic of this disorder is that bodily secretions become viscous and dry out, causing clogging of airways and pores of organs such as the lungs, the pancreas and the liver. Main clinical features include progressive pulmonary obstruction, pancreatic failure, malabsorption, male infertility and a decreased life expectancy. The disease is expressed and inherited as an autosomal recessive genetic disorder, which means that affected individuals must have mutations in both copies of the CFTR gene, typically inherited from both carrier parents.
Especially in males with reproductive problems, what is of particular importance is that a specific type of mutations in the CFTR gene lead to congenital bilateral absence of the vas deferens (CBAVD), with clinical symptoms of obstructive azoo/oligospermia. CBAVD is responsible for ~1.5% of male infertility in general and ~ 80% of patients with CBAVD harbor at least one mutation in the CFTR gene. Thus, in men with obstructive azoospermia, and especially in the context of preparation for IVF, the possible existence of cystic fibrosis mutations should be investigated specifically in order to confirm this type of abnormality, through a specially adapted genetic analysis. Furthermore, genetic testing in this context is also important, since if a mutation is detected in the male partner and due to the high frequency of disease carriers in the population (1 in 25), immediate genetic testing of the female partner should follow, through extended testing for> 99% of mutations, in order to determine the couple’s risk for having an affected child.
There are two particular issues to consider in genetic testing for cystic fibrosis:
- mutation carriers are perfectly healthy, with no clinical symptoms and therefore not easily recognizable, and
- the CFTR gene is relatively large in size with many possible mutations (> 1800)
Επιπλέον, οι μεταλλάξεις στο γονίδιο δεν είναι ίδιες σε συχνότητα σε όλους τους πληθυσμούς, με πολύ σημαντικές διαφορές, για παράδειγμα, μεταξύ λαών της Β. Ευρώπης και της Μεσογείου. Λόγω λοιπόν του μεγάλου αριθμού, αλλά και της διαφορετικής συχνότητας των μεταλλάξεων στους διάφορους πληθυσμούς-εθνότητες, είναι σχεδόν αδύνατο να σχεδιασθεί μια συγκεκριμένη ομάδα-αριθμός μεταλλάξεων που θα ελέγχονται παγκοσμίως. Η ανάλυση για την αποκάλυψη φορέων της νόσου συνιστάται να εκτελείται για ποσοστό που να καλύπτει ~85% των μεταλλάξεων του συγκεκριμένου πληθυσμού.
Extended molecular genetic testing for cystic fibrosis is indicated for confirmation of clinical diagnosis, in high-risk cases, for the detection of rare mutations and in male infertility due to obstructive azoospermia.
It should be noted that according to international guidelines and recommendations, and regardless of the cost, extended genetic testing of the entire CFTR gene is not recommended as a routine test for carrier detection in the population, as it very often reveals mutations which are not pathogenic, thus leading to unwarranted anxiety and unnecessary further tests.
We perform DNA sequence analysis, via Next Generation Sequencing (NGS) on a Genome Analyzer – Ion Proton platform, of all exons and intron-exon junctions/splice sites of the CFTR gene, coupled to MLPA analysis for the detection of deletions/duplications, thus detecting >99% of all pathogenic mutations of the disease.
InterGenetics participates with great success in the external quality assessment scheme organized by the European Molecular Genetics Quality Network (EMQN) – CF Network, which is periodically applied for Cystic Fibrosis.