Το σύνδρομο Apert ανήκει στην ομάδα των συνδρόμων κρανιοσυνοστεώσεων κι εκδηλώνεται με κρανιοεγκεφαλικές δυσπλασίες, συνδακτυλία στα άνω και κάτω άκρα και τήξη στη δομή των οστών (με πιθανή παρουσία ενδιάμεσων μαλακών ιστών). Το σύνδρομο Apert έχει το μεγαλύτερο κίνδυνο για ήπια και μέτρια διανοητική καθυστέρηση από όλα τα κρανιοσκελετικά σύνδρομα.
Σύνδρομο Apert
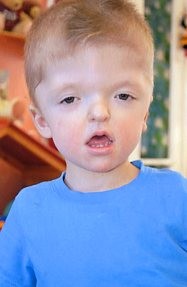
Όπως και τα υπόλοιπα σύνδρομα αυτής της ομάδας, είναι ένα αυτοσωματικό επικρατές νόσημα, αν και η πλειοψηφία των περιπτώσεων είναι σποραδικές. Προκαλείται απο μεταλλάξεις στο γονίδιο FGFR2, οι οποίες στην πλειοψηφία τους συμβαίνουν στα εξόνια 8 και 10 του γονιδίου. Η προχωρημένη αναπαραγωγική ηλικία του πατέρα έχει κλινικά αποδειχθεί ότι συνδέεται με de novo μεταλλάξεις των συνδρόμων Crouzon, Pfeiffer, Beare-Stevenson και Muenke.
Σε κάθε περίπτωση, ο προγεννητικός έλεγχος για τα περισσότερα σύνδρομα κρανιοσυνοστεώσεων δεν είναι διαδεδομένος κι αποτελεί ευαίσθητο θέμα, αφού τα σύνδρομα αυτά θεραπεύονται με κατάλληλη διορθωτική χειρουργική επέμβαση.
Στο εργαστήριο μας πραγματοποιούμε ανάλυση της αλληλουχίας του DNA (automated bi-directional fluorescent DNA sequencing) των εξονίων 8 και 10 του γονιδίου FGFR2 , επιτρέποντας την ανίχνευση ποσοστού μεγαλύτερου από 90% των παθολογικών μεταλλάξεων του νοσήματος.