Το σύνδρομο Smith-Lemli-Opitz προκαλείται από υπολειπόμενου τύπου μεταλλάξεις στο γονίδιο DHCR7 στη χρωμοσωματική θέση 11q12, που κωδικοποιεί το ένζυμο 7-dehydro-cholesterol reductase, που οδηγούν σε ανωμαλίες του μεταβολισμού της χοληστερόλης και ανεπάρκεια των παραγώγων στεροειδών ορμονών.
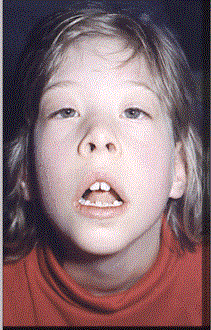
Οι φαινοτυπικές ανωμαλίες περιλαμβάνουν μικροκεφαλία, υποσπαδία, αμφίβολα εξω-γεννητικά όργανα, διανοητική καθυστέρηση και δυσπλασίες του προσώπου. Μερικά απο αυτά τα χαρακτηριστικά μπορεί να αποκαλυφθούν κατά τη διάρκεια της προγεννητικής υπερηχογραφικής εξέτασης (π.χ. υποσπαδία ή/και κρυψορχία) κι επειδή τα χαμηλά επίπεδα ορού της ελεύθερης οιστριόλης στον ορό της εγκύου μητέρας επίσης μπορεί να συνδέονται με το νόσημα, η ανάγκη προγεννητικού ελέγχου για μεταλλάξεις του γονιδίου DHCR7 στο έμβρυο είναι σχετικά συχνό γεγονός.